NUCYNTA: WELL-DEFINED TOLERABILITY1
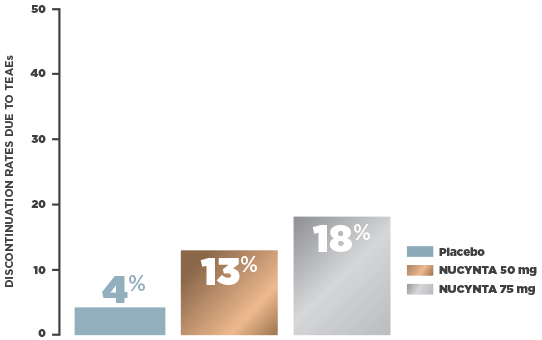
- Safety population (n=666): All subjects with end-stage degenerative joint disease receiving at least 1 study drug intake following randomization
Oxycodone IR was included in the study as an active control.1
AEs=adverse events.
Study design:
In a multicenter, randomized, double-blind, active‑ and placebo‑controlled, parallel group, phase 3 study, subjects who were candidates for joint replacement surgery due to end‑stage joint disease (n=659, ITT) were selected to assess the efficacy and safety of NUCYNTA. Patients were randomized 1:1:1:1 to receive NUCYNTA 50 mg, 75 mg, oxycodone IR 10 mg, or placebo. Patients received study drug every 4 to 6 hours during waking hours for 10 days. The primary efficacy endpoint was the sum of pain intensity difference (SPID) over 5 days.1
NUCYNTA: WELL-DEFINED TOLERABILITY1,2
ADVERSE REACTIONS REPORTED BY ≥1% OF NUCYNTA-TREATED PATIENTS IN SEVEN PHASE 2/3 PLACEBO- AND/OR OXYCODONE-CONTROLLED, ONE NON-CONTROLLED, AND ONE PHASE 3 OXYCODONE-CONTROLLED SAFETY, MULTIPLE-DOSE CLINICAL STUDIES3

References:
- Hartrick C, Van Hove I, Stegmann J-U, et al. Efficacy and tolerability of tapentadol immediate release and oxycodone HCI immediate release in patients awaiting primary joint replacement surgery for end-stage joint disease: a 10-day, phase III, randomized, double-blind, active- and placebo-controlled study. Clin Ther. 2009;31(2):260-271.
- Data on file. Collegium Pharmaceutical, Inc. (EDJD).
- NUCYNTA (package insert). Stoughton, MA: Collegium Pharmaceutical, Inc; December 2023.